Transitioning bioprocesses from R&D to full-scale production poses intricate challenges, especially in advanced modalities like cell and gene therapy. Access to high-quality equipment and materials that can support processing from the lab to the production plant can help reduce the complexity of scale-up and the associated risks. Corning Life Sciences, with its emphasis on science, innovation, and customer collaboration and its comprehensive portfolio of equipment, consumables, and ancillary materials, has long been a mainstay in the laboratory and is ideally positioned to help cell and gene therapy manufacturers bring their curative treatments to market.
Biologic, Sourcing, and Regulatory Challenges to Scaling
Scaling presents many challenges in biologics manufacturing, regardless of the type of drug substance involved — conventional monoclonal antibody, next-gen multispecifics, or novel modalities, such as viral vectors and cell therapies.
The first challenge is rooted in the fundamental biology. Process conditions in compact shake flasks vary considerably from those in expansive stirred-tank bioreactors. Factors, such as mixing dynamics, gas concentrations, nutrient and metabolite levels, and pH values across the medium, all undergo changes when transitioning from the laboratory bench to the pilot stage and eventually to full-scale commercial production. Adapting to these variations demands intricate process comprehension and, ideally, the implementation of scaled-down production models to bridge the differences in process parameters.
The second obstacle relates to the selection and procurement of raw materials. Regulatory bodies are placing heightened emphasis on the quality of even ancillary materials, which might not be present in the final product, recommending their production under GMP conditions. R&D labs typically operate at smaller scale; many of the materials they use may be designated for research use only (RUO) and may not always meet the highest quality standards. As their process scales up, the challenge arises when a vendor does not offer a GMP-grade variant of that product with consistent properties; transitioning to an acceptable alternative can then affect both the process performance and the final product quality. This issue can be mitigated substantially by initially selecting raw materials, such as Corning® CellSTACK® cell culture vessels, that are available with consistent characteristics in both research and GMP scales.
A third distinct challenge in bioprocess scaling stems from the need to align with the regulatory standards of the markets where the biopharmaceutical product is intended for sale. Beyond raw material standards, there are requirements for product quality and process monitoring and control. The transition into clinical and commercial production is more than just a question of biology and sourcing — it encompasses how every facet of bioprocessing converges at a larger scale to yield drug substances and products that ensure safety and efficacy for the treatment of human disease.
The Scale-Out vs. Scale-Up Question
Another pivotal consideration for manufacturers when producing larger volumes is deciding between scaling up and scaling out. It is crucial to determine the required, phase-appropriate quantities of drug substance and product and the most effective technology to meet those needs. There is a marked distinction between preclinical, phase I clinical, phase III clinical, and full commercial production stages, and the ultimate scaling approach is typically determined by the end of phase II or before phase III.
Scaling out means amplifying output by replicating the process using identical equipment, as many times as needed to meet demand. This method utilizes consistent technology, eliminating the need for additional process optimization. However, plant footprint, human resources (number of operators), and timing are critical considerations.
Increasing the output through scaling up typically requires transitioning to larger-volume or higher-surface processes, often using more expansive equipment. This approach allows the same team of operators to produce significantly more product, but more process development work is required to ensure that the process is optimized for the larger scale. This effort can be minimized by using production systems designed to be scalable, such as Corning CellSTACK, and HYPERStack® cell culture vessels, which provide similar process conditions for adherent cell culture and thus streamlined scale-up.
In some cases, scaling up not only means producing a greater volume but also transitioning between technologies. For example, viral vector manufacturing via transient transfection typically employs adherent cell culture at smaller scales. Yet, as production processes expand, manufacturers often shift toward utilizing suspension cell culture.
An alternative approach is to keep adherent cells in a suspension process utilizing Corning’s polystyrene or dissolvable microcarriers within a single-use bioreactor (SUB). Alternatively, the Corning CellCube® system provides a perfusion-based method in which cells are attached to both sides of layered plates within enclosed modules of 10, 25, and 100 layers. These methods facilitate significant scaling within a compact footprint.
Indeed, many pharmaceutical manufacturers strategically leverage both scaling out and scaling up, depending on the need. For instance, antibody production is often scaled to 2,000-L SUBs. When demand surpasses this scale’s capacity, instead of transitioning to larger 5,000-L bioreactors, manufacturers typically choose to scale out using 6 x 2,000-L bioreactors. This strategy circumvents additional process development and reduces risks; if one batch out of the six fails, the other five remain unaffected. This approach not only minimizes economic setbacks but also crucially prevents potential drug shortages and risks to patients.
The Need to Align Numerous Moving Parts
Successful scale-up and transfer of processes from one team to another, each with different objectives and workflows, requires alignment of many different moving parts. R&D environments often contrast sharply with manufacturing settings. The former tend to operate with more flexibility, while the latter emphasize strict control, influencing all aspects of bioprocessing. Operators oversee and observe processes, often equipped with inline sensors and automated mechanisms for media and feeds. Within a cleanroom setting, these operators are clad in full bodysuits. Furthermore, the time required to process a large-scale batch far exceeds that of a small-scale operation.
Before transitioning technology from R&D to a manufacturing setting, it is vital to account for the numerous differences between these environments. While it is standard to have teams focused on tech transfer and scaling, that alone is not necessarily adequate. R&D teams need to understand the varied approaches and workflows, aiming to adapt the biology to ensure that processes are robust and scale seamlessly.
Engaging in discussions about whether to scale up or out at the earliest stages is crucial to ensure comprehensive consideration throughout process development.
Focus on Increasing Sustainability
Corning Life Sciences, along with the entire pharmaceutical industry, recognizes the need to increase the sustainability of drug manufacturing operations. In response, Corning is rolling out a number of sustainability initiatives, including the Corning® EcoChoice™ program. Products under the Corning EcoChoice label are manufactured, packaged, and distributed in a manner that prioritizes environmental responsibility, adhering to the FTC guidelines, which require all sustainability statements to be specific, evidence-based, and traceable. Strategies employed to increase sustainability include use of recycled materials, source reduction, process intensification, and use of renewable energy.
Corning HYPERFlask® and HYPERStack Vessel Case Studies[1]
In the cell and gene therapy sector, scaling up from initial R&D to larger-scale adherent cell culture is a common challenge. Corning Life Sciences recognized this challenge and established a bioproduction division, offering support through class one medical devices like the Corning HYPERFlask, CellSTACK, and HYPERStack products. Corning’s extensive materials science expertise ensures uniformity in raw materials, aiding a smooth transition during scale-up and preventing variations that can affect cell adhesion and growth. Corning’s GMP-compliant technologies range from traditional spinner flasks to advanced flaskware and dissolvable microcarriers and extends to the Ascent® Fixed Bed Bioreactor and CellCube technology to accommodate projects at various developmental stages and scales. Additionally, Corning provides a comprehensive suite of cell culture solutions, streamlining upstream processes for cell and gene therapies. Our commitment includes collaboration with customers throughout all project stages, integrating automation, adhering to regulatory standards, and gathering insights to address a host of production challenges.
For example, one customer looking to bring production in-house to supplement production at a CDMO needed a solution that would fit in an existing space and provide specific incubation conditions. Corning HYPERStack vessels were found to be the ideal solution, optimizing the utilization of their existing manufacturing space.
In another case, research scientists at a large biopharmaceutical company found themselves overwhelmed with managing hundreds of T-flasks. Seeking alternatives, they approached Corning for a solution. While the project is ongoing, the preliminary transition sees those numerous T-flasks substituted with merely 10–15 HYPERFlask vessels.
Additionally, during a phase I trial, the Ottawa Hospital Research Institute’s Cell Manufacturing Facility needed to deliver freshly cultured allogeneic bone marrow–derived MSCs to septic shock patients within a narrow 6-hour window. The Corning HYPERFlask vessel was pivotal for this emergent per-patient dosing. As the research advanced to larger and later-phase clinical trials, the consistent and scalable nature of the Corning HYPERFlask and HYPERStack cell culture vessels ensured reproducibility, maintaining the same protocol and working environment of the isolator units.
iPSC Opportunities
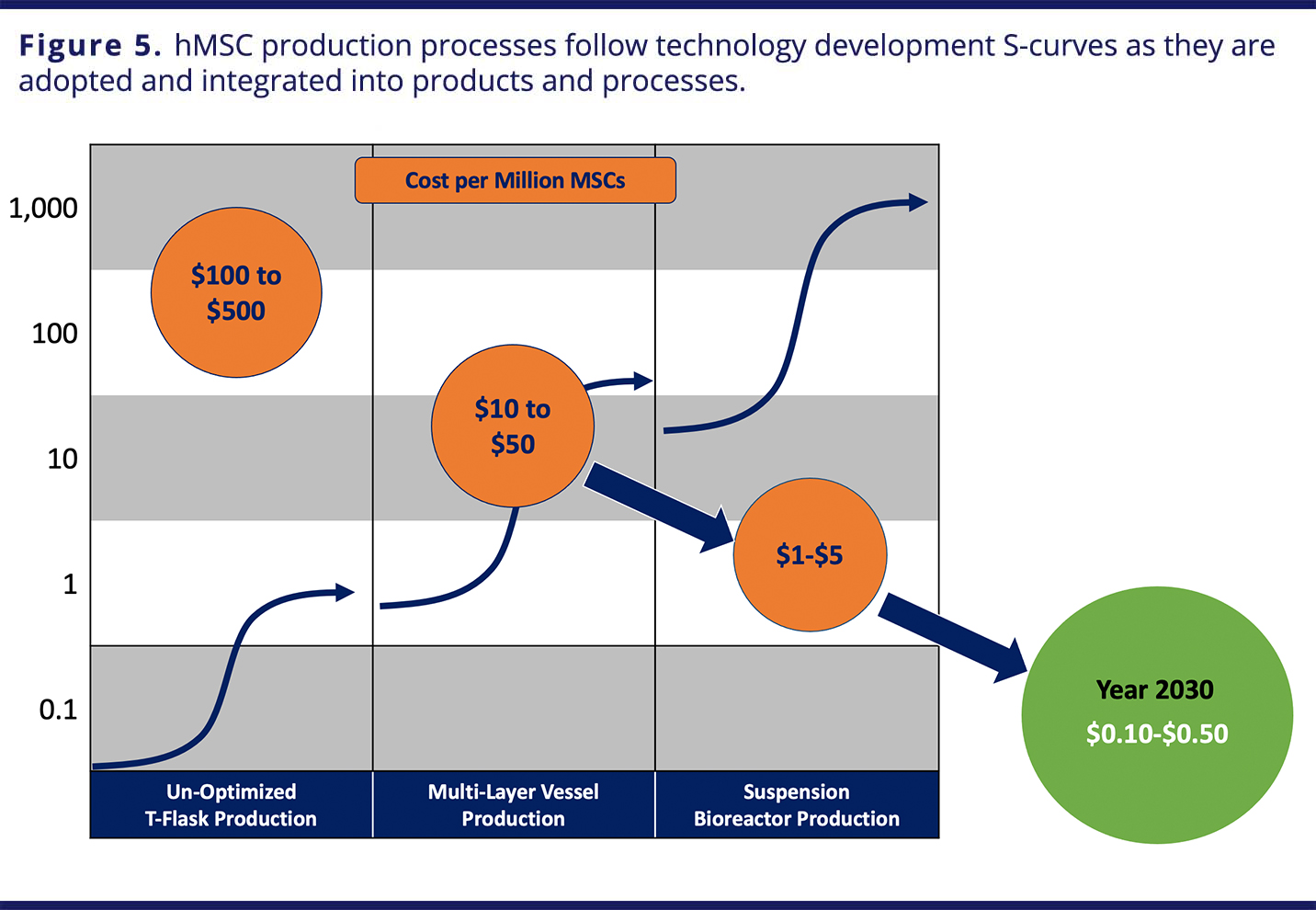
Stem cells, especially induced pluripotent stem cells (iPSCs) and mesenchymal stem cells (MSCs), are gaining traction in non-genetically modified cell therapies. Traditionally, these cells have been cultivated in a 2D adherent culture. There is growing interest in suspension culture, due to its promise of greater scalability and cost-efficiency. However, moving to suspension presents challenges, particularly for stem cells, which prefer adherent environments. Microcarriers — small beads that provide a high ratio of surface area to volume — suspended in SUBs offer an alternative approach with some of the benefits of both adherent and suspension environments, and dissolvable microcarriers (DMCs), like Corning’s Synthemax® II DMCs, enable a more efficient harvest and downstream experience.
Innovation Driven by Science
Cell and gene therapies signify a transformative approach in medical science, shifting from merely managing symptoms to achieving complete cures for patients. Corning Life Sciences stands committed to realizing this transformative vision alongside its partners. To make that happen, the company values science and strives to innovate in collaboration with customers. As partners pave the way with novel products and methods, Corning harnesses its scientific prowess to deliver cutting-edge equipment and material solutions, propelling these groundbreaking cell and gene therapies into the mainstream market.
Originally published on PharmasAlmanac.com on December 12, 2023.